 Ke Lu
Ke Lu Wen Wang1†
Wen Wang1† Jiye Liu
Jiye Liu Lijie Xing
Lijie Xing
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 17 July 2024
Sec. Hematologic Malignancies
Volume 14 - 2024 | https://doi.org/10.3389/fonc.2024.1413494
Multiple myeloma (MM) is the most prevalent malignant monoclonal disease of plasma cells. There is mounting evidence that interactions with the bone marrow (BM) niche are essential for the differentiation, proliferation, survival, migration, and treatment resistance of myeloma cells. For this reason, gaining a deeper comprehension of how BM microenvironment compartments interact with myeloma cells may inspire new therapeutic ideas that enhance patient outcomes. This review will concentrate on the most recent findings regarding the mechanisms of interaction between microenvironment and MM and highlight research on treatment targeting the BM niche.
Multiple myeloma (MM) is characterized by clonal expansion of malignant plasma cells within the bone marrow (BM), leading to organ failure at the time of diagnosis, which includes calcium elevation, renal dysfunction, anemia, and/or bone disease (CRAB) (1). Remarkable discoveries in the biology of MM have transformed the treatment paradigm and extended median patient survival over the past two decades. Nonetheless, MM remains mostly incurable due to genetic complexity and instability, as well as the permissive, tumor-promoting BM microenvironment. Thus, new therapeutics targeting both myeloma cells and immunosuppressive microenvironment are urgently needed.
The BM microenvironment is a heterogeneous system comprising a cellular compartment (e.g., immune cells (including myeloid-derived suppressor cells, dendritic cells, macrophages, T-cells, natural killer cells, regulatory B cells), osteoblasts, osteoclasts, endothelial cells, and stromal cells) and a non-cellular compartment [e.g., extracellular matrix (ECM), extracellular vehicles (EVs), oxygen concentration, and the liquid milieu (cytokines, growth factors, and chemokines)] (Figure 1). The interaction between myeloma cells and the BM milieu encourages tumor immune escape and promotes the former’s proliferation, survival, dissemination, and drug tolerance via a variety of mechanisms (2–4). In this review, we describe the mechanisms by which each BM milieu member promotes the development of MM cells and outline potential therapies to target them.

Figure 1 Bone marrow microenvironment of multiple myeloma.
MDSCs are a diverse population of immature myeloid cells (IMCs) that, under normal conditions, can differentiate into granulocytes, macrophages, or dendritic cells under steady-state settings (5). However, in pathological conditions typical of malignancies, the differentiation of IMCs was reported to be inhibited, resulting in the accumulation of MDSCs. Current studies identify two main subtypes of MDSCs: monocytic myeloid-derived suppressor cells (M-MDSCs), which are phenotypically and morphologically similar to monocytes, and granulocytic/polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs), which are phenotypically and morphologically similar to neutrophils. A significant presence of MDSCs is found in the multiple myeloma tumor microenvironment, where they play a crucial role in immune escape and disease progression (6, 7). It has become clear that there is a bidirectional interaction between MM cells, MDSCs, and immune effector cells. It has been demonstrated that M-MDSC plays a more predominant immunosuppressive role in the multiple myeloma tumor microenvironment. As a precursor of osteoclasts, M-MDSC contribute to bone destruction, leading to severe bone pain or pathological fractures in some MM patients. Conversely, PMN-MDSCs are more involved in neovascularization mechanisms within the bone marrow microenvironment (6, 8). MDSCs exert immunosuppressive effects through multiple mechanisms: they produce and release inhibitory cytokines and soluble factors such as IL-10 and TGF-β, and induce regulatory T cells (Tregs) (9, 10); they generate reactive oxygen species (ROS), which disrupt the ability of CD8+ T cells to bind to peptide-major histocompatibility complexes (11). MDSCs also reduce tryptophan levels in the tumor microenvironment due to the expression of Arg1 and Indoleamine 2,3-Dioxygenase (IDO) enzyme activity, which diminishes TCR formation, inhibits T-cell proliferation and induces T-cell cycle arrest by depletion of L-arginine (12, 13). Additionally, MDSCs can increase the levels of anti-apoptotic proteins in MM cells by inducing AMPK phosphorylation, thereby enhancing the proliferation of MM cells (14). They also endow stem-like qualities to myeloma cells, promoting epithelial-mesenchymal transition (EMT) (15). Conversely, malignant plasma cells stimulate the growth of MDSCs (16).
DCs are expert antigen-presenting cells (APCs) that deliver antigens to T cells and, depending on their functional condition, either induce immunity or tolerance. Two main subgroups of DCs can be distinguished by their origin, phenotype, and function: myeloid DCs (mDCs) and plasmacytoid DCs (pDCs) (17). In the literature, there is no universal consensus on the phenotype, function, and frequency of DC population in MM patients compared to healthy individuals. Nonetheless, considerable research has shown that DCs in MM patients’ BM are dysfunctional, resulting in diminished anti-tumor immune responses and myeloma escape (18–20). It has been discovered that interactions between DCs and MM cells can confer proliferation, survival, and medication resistance against tumor cells through receptor activator of nuclear factor-κB (RANK)/RANK ligand (RANKL) signaling and a proliferation-inducing ligand (APRIL)-mediated interactions (21), as well as CD28 and CD80/CD86 crosslinking (22, 23). In addition, pDC-MM interaction triggers the secretion of cytokines and chemokines, which not only helps MM cells grow, survive, and develop treatment resistance, but also extends the survival of pDCs (24). More recently, Ray et al. used next-generation sequencing (NGS) to examine genomic alterations in MM cells induced by co-culture with pDCs. They discovered that co-culturing pDCs with myeloma cells increases CD73, CD274, HDAC6, TLR7/9, or IL3Ra/CD123 gene expression, while reducing the expression of ADAM33, BAD, BAK1, and CASP3 in tumor cells. These results make it possible to induce MM-specific CD8+ CTL activity by blocking CD73. Furthermore, combining an anti-CD73 antibody with a TLR7 agonist significantly increases the cytotoxic activity of MM-specific CD8+ CTL (25). Altogether, these findings highlight the importance of DCs in the etiopathogenesis of MM and present novel therapeutic targets to improve patient outcomes.
Macrophages are known as specialized phagocytic cells that are essential for both innate immune response and tissue repair. There are two main subsets of macrophages: M1 type macrophages (M1) and M2 type macrophages (M2). In the tumor setting, M1 typically acts as a potent anti-tumor effector and kills tumor cells by mediating direct cytotoxicity and antibody-dependent cell-mediated phagocytosis (ADCP). In contrast, M2 facilitates tumor growth, invasion, metastasis, and treatment resistance (26). Tumor-associated macrophages (TAMs) originate from circulating monocytes, which are recruited to the tumor site by cytokines released by tumor cells (27, 28). In comparison to Monoclonal Gammopathy Of Undetermined Significance (MGUS), MM exhibits higher expression of TAMs, suggesting their potential role in the transition from MGUS to MM (29, 30). Previous research indicates that during disease progression, the tumor microenvironment in MM undergoes macrophage reprogramming. Reprogrammed TAMs demonstrate a mixed phenotype, displaying characteristics of both M1 and M2 macrophages, with a predominant M2-like profile. These reprogrammed TAMs are associated with impaired phagocytic function (30, 31). Multiple myeloma cells recruit macrophages that support tumor growth and encourage their polarization toward an M2-like phenotype via several different mechanisms (29, 32–34). TAMs exert multifaceted effects on MM, influencing proliferation, migration, angiogenesis, immunosuppression, and drug resistance (35). They promote MM cell proliferation by upregulating secretion of IL-6 and IL-10 (36–38), and enhance migration by inducing cytokine-mediated vascular leakage and downregulating CD138 and C-X-C motif chemokine receptor 4 (CXCR4), thus reducing cell adhesion (39–41). TAMs contribute to angiogenesis both directly and indirectly: they produce and release angiogenic factors such as vascular endothelial growth factor (VEGF) and matrix metalloproteinases (MMPs), and in vitro, they cooperate with MM cells to stimulate proliferation, migration, and tubule formation of Human Umbilical Vein Endothelial Cells (HUVECs). Additionally, TAMs exposed to VEGF and basic fibroblast growth factor can directly form capillary-like blood vessels by acquiring endothelial cell markers (42–44); Moreover, TAMs induce immunosuppression by downregulating IFN-γ, inhibiting MHC Class II molecule expression to limit effector T cell function, and modulating immunosuppression through the macrophage immune checkpoint CD47-SIRPα (29, 41, 45, 46); Studies also indicate that TAMs contribute to increased drug resistance in MM cells (47, 48). To sum up, MM cells can manipulate macrophages to facilitate tumor settlement and progression. Accordingly, anti-TAM therapeutic strategies are emerging as intriguing and promising approaches. Much evidence indicates that TAMs contribute to MM cell resistance to chemotherapeutic drugs. TAMs can mediate bortezomib resistance by secreting IL-1β, which increases the number of MM-tumor-initiating cells (49). Additionally, TAMs can express B-cell activating factor (BAFF), preventing bortezomib-induced apoptosis through the classical and alternative NF-κB pathways (50). Moreover, TAMs impact the efficacy of CAR-T therapy. A report showed that in B cell NHL, patients achieving complete responses with CAR-T therapy had decreased levels of TAMs, Treg cells, and MDSCs, whereas chemokines and MDSCs were overexpressed in patients achieving only partial remission (51).
The progression of MM is associated with a deteriorating innate and adaptive immune system, particularly affecting the T-cell repertoire. CD8+ cytotoxic T lymphocytes (CTLs) mediated cytotoxicity plays a pivotal role in anti-tumor T-cell responses (52). However, T-cell dysfunction has been observed even in individuals at precursor stages of plasma cell dyscrasia, such as increased levels of T-cell exhaustion in monoclonal gammopathy of undetermined significance (MGUS) (53) and reduced expression of activation markers in smoldering multiple myeloma (SMM) (54). Notably, T-cell function progressively deteriorates throughout the disease course. In MM patients, especially those with relapsed/refractory MM (RRMM), co-inhibitory molecules including PD-1 and TIGIT are upregulated on activated T-cells, which protects myeloma cells from immune attack by directly interacting with their ligand expressed in myeloma cells (55–57). However, up to now, immune checkpoint inhibitors (ICIs) have failed to show promising clinical benefits in MM, possibly due to compromised antigen-specific T-cell function or loss of stem-like/tissue-resident memory T (TRM) cells (53, 58).
CD4+ T-cells can be categorized into several functionally distinct T-cell subsets: T helper 1 (TH1), TH2, TH17, TH22, T follicular helper (TFH), and regulatory T-cells (Tregs) (59). The specific immune imbalance of CD4+ T-cell subsets on MM remains unclear and controversial. Nevertheless, several research findings are more widely accepted. Firstly, TH1 cells collaborate with tumor-infiltrating, antigen-presenting macrophages to achieve anti-tumor responses (60). Then, MM cells induce the generation of Tregs via multiple mechanisms (61–65), contributing to immune dysfunction and negatively influencing clinical outcomes (66). Lastly, in MM patients, there is a significant increase in TH17 cells and related cytokines, such as IL-17, which promotes MM cell proliferation while inhibiting immunological responses (67–70). Strikingly, TH2 cells, which are generally considered to have a pro-tumorigenic role, have been shown to eradicate myeloma by triggering an in situ inflammatory immune response (71).
In recent years, unconventional T cells have garnered increased attention for their pivotal role in hematological tumors. Among these, γδ T cells, MAIT cells, and iNKT cells (invariant natural killer T cells) exhibit both innate and adaptive immune characteristics, primarily characterized by their rapid recognition of unconventional peptide antigens. Stimulated γδ T cells demonstrate potent cytotoxic effects on MM cells in vitro (72), with Vδ1 cells being activated by various receptors, including T-cell receptors and molecules such as NKG2D, CD3, CD2, DNAX accessory molecule-1, and intracellular adhesion molecule-1 (ICAM-1). This activation confers cytotoxic capabilities against MM cells, suggesting a potential therapeutic strategy for MM (73). Additionally, studies have indicated a reduction in the percentage of MAIT cells in MM patients compared to healthy individuals, accompanied by decreased production of IFN-γ and reduced CD27 expression in MAIT cells at disease onset, indicative of MAIT cell depletion (74).
Natural killer (NK) cells are vital to the innate immune response due to their direct cytotoxic activity and antibody-dependent cellular cytotoxicity (ADCC). Previous studies have shown that NK cell reactivity is mediated by the expression of a wide variety of inhibitory receptors characterized by CD94-NKG2A heterodimeric receptors, killer cell Ig-like receptors (KIRs), as well as activating receptors such as CD16, NKG2D, DNAM-1, activating KIR, and the natural cytotoxicity receptor (NCR) family (75). Malignant plasma cells can evade NK cell-mediated killing by downregulation or blocking activating receptors and activating Tregs or regulatory B cells (Bregs) (76–79).
The potential effects of B-cell subsets on the BM milieu in MM are not well characterized. However, emerging research suggests a small immunosuppressive B-cell subset known as Bregs has the ability to mediate evasion of myeloma plasma cells from the immune system. Malignant plasma cells in the bone marrow support the survival of Bregs by preventing their apoptosis and activating the APRIL/TACI axis (63, 79). Consequently, Bregs promote an immune suppressive microenvironment through the production of IL10 and alternative mechanism including the interference with NK cell-mediated ADCC against MM cells. Thus, Bregs are a promising new therapeutic target in MM.
In a steady state, the differentiation and activity of OCs and OBs maintain a balance between bone resorption and formation, ensuring bone homeostasis and integrity. In MM environment, the OC-OB axis is disrupted leading to increased bone resorption and impaired bone formation, ultimately resulting in osteolytic bone disease. In MM, tumor PCs produce both activators of OCs and inhibitors of OBs (80): MM cells increase the differentiation and activity of OCs by dramatically increasing the release of osteoclastogenic factors (81–87) and favoring the recruitment of OCs precursors (88–90), conversely, malignant plasma cells can elevate Dickkopf-1 (DKK1) levels (91) and decrease the essential OB transcription factor RUNX2 activity (92) leading to the suppression of osteoblast activity and osteoblast differentiation. OBs play a crucial role in bone formation by producing bone morphogenetic protein (BMP), which binds to receptor and activates downstream osteoblast-specific transcription factor RUNX2,OSTERIX, etc (93). Additionally, Wnt in OBs binds to LRP-5/6, activates β-catenin and promotes the differentiation of bone marrow mesenchymal stem cells to osteoblasts while inhibiting their apoptosis (94). Sclerostin (SOST), primarily secreted by osteoblasts, serves as a significant negative regulator of bone formation. Elevated levels of SOST expression hinder the Wnt and BMP signaling pathways, thereby impeding osteoblast differentiation and proliferation and ultimately inhibiting bone formation (95). Importantly, recent studies reveal that the function of OCs and OBs not only contributes to bone-remolding, but also involves maintaining an immunosuppressive myeloma environment. OCs shield myeloma cells from T-cell cytolytic function via high expression of checkpoint molecules including PD-L1, IDO HVEM, CD200, and Galectin-9 (96). Myeloma-osteoclast interactions upregulate Chondroitin synthase 1 (CHSY1), triggering Notch signaling and boosting tumor cell proliferation and bone resorption (97). Additionally, through the modification of the endosteal niche, OCs are able to control the resurgence of dormant myeloma cells (98, 99). Lastly, OCs encourage angiogenesis, which is necessary for the survival and multiplication of MM cells (100, 101). As for OBs, Khoo WH et al. have found that the interactions between MM and osteoblasts may also play a role in maintaining tumor cell dormancy (102). Taken together, the above findings underline the therapeutic value of targeting OCs to modulate BM microenvironment, improve anti-tumoral immune responses, and improve the bone phenotype.
Tom Cupedo et al. conducted a comprehensive mapping of the myeloma inflammatory stromal microenvironment using single-cell transcriptome sequencing. They identified myeloma-specific inflammatory stromal cells that spatially colocalize with tumor and immune cells, are induced by inflammatory factors, and provide essential survival factors to plasma cells. The MM microenvironment was characterized by the expansion of IFN-responsive T cells, CD8+ Tscm cells and GZMK+CX3CR1-CD56bright NK cells, while the distribution of other T cells, B cells and mononuclear myeloid cells remain unaltered (103). Additionally, Tom Cupedo and colleagues reported that neutrophils in the bone marrow of MM patients are activated to promote the transcription of IL-1β and myeloma cell survival factor TNFSF13B (BAFF). These neutrophils establish a positive feedback loop with inflammatory stromal cells, thereby perpetuating a tumor-supportive inflammatory environment after treatment (104). BMSCs and vascular endothelial cells can produce a chemoattractant called stromal cell-derived factor 1 alpha (SDF-1a). The binding of SDF-1a to its receptor CXCR4 is involved in the mobilization and homing of hematopoietic stem and progenitor cells (105). In MM, myeloma cells utilize CXCR4 to interact with SDF-1a, resulting in the adhesion of myeloma cells to BMSCs and endothelial cells. This interaction leads to the overexpression of factors such as vascular endothelial growth factor (VEGF), hepatocyte growth factor (HGF), IL-6, IL-3, and tumor necrosis factor alpha (TNFα) which contributes to osteolysis, angiogenesis, as well as MM cell survival and proliferation (106). The binding of malignant plasma cells to BMSCs also triggers the activation of various adhesion molecules such as very-late-antigen-4 (VLA-4) and CD44, which mediates cell adhesion-mediated drug resistance (CAM-DR) (107). Recent studies revealed that CXCR4 could enhance the acquisition of epithelial-to-mesenchymal transition (EMT-like) phenotype in MM cells, promoting extramedullary disease (EMD) development both in vivo and in vitro (40, 108).
The BM extracellular matrix (ECM) consists of proteins such as fibronectin, collagen, osteopontin, hyaluronan, and laminin, serving as a scaffold for tumor cells to cling to and interact with its components or other cells. MM cells directly interact with the ECM by binding integrins like VLA-4 and integrin b7 (ITGB7) to ECM proteins. This interaction is required for MM cell survival and contributes to CAM-DR (109–112). Besides, CD138 expressed on MM cells binds to type I collagen and promotes matrix metalloproteinase 1 (MMP1) production, thereby stimulating tumor invasion, bone resorption, and angiogenesis (113, 114).
Another important component of BM microenvironment is extracellular vehicles (EVs). EVs are a diverse class of membranous structures released by all kind types of cells and act as important intermediaries between myeloma cells and the surrounding milieu or remote premetastatic niche (115). Recent researches have uncovered the crucial role of EVs in tumor progression through various mechanisms, including but not limited to angiogenesis moderation (116–118), participation in the formation of bone lesions (119–122), mesenchymal cell education (123–126), and immune modulation (127–130).
In addition, it has been demonstrated that the BM of MM mouse models and MM patients is hypoxic compared to healthy controls (131, 132). The decrease in oxygen concentration enhances the acquisition of stem cell-like or EMT-like phenotype in MM cells, thus enhancing their dissemination properties (133, 134). Also, hypoxia contributes to the progression of osteolytic bone disease as well as angiogenesis (135, 136). Targeting the hypoxia microenvironment should be considered as a novel anti-MM treatment strategy and has the potential to synergize with other anti-MM therapies (137).
MM treatments targeting the BM microenvironment aim to disrupt the complex interactions between MM cells and the surrounding environment and restore certain cellular functions, altering the supportive environment for MM cells. By understanding the molecular and cellular changes that occur in the BM microenvironment in response to MM, the drugs are selected or designed to target specific components and pathways that are affected by MM (Figure 2). Multiple drugs have shown efficacy in interfering with MM cell growth, survival, and drug resistance (Table 1).
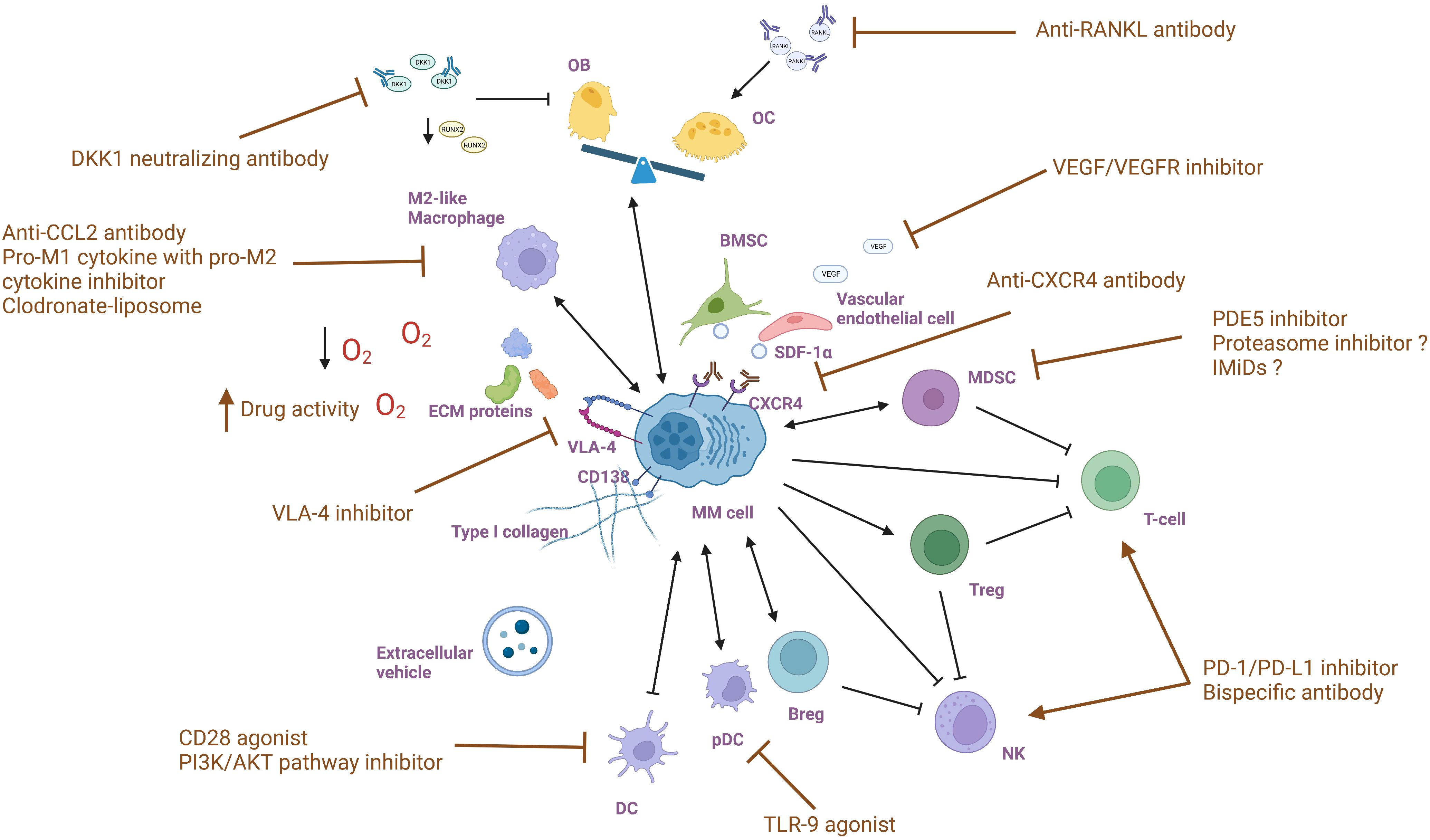
Figure 2 Targeting bone marrow microenvironment components in multiple myeloma.
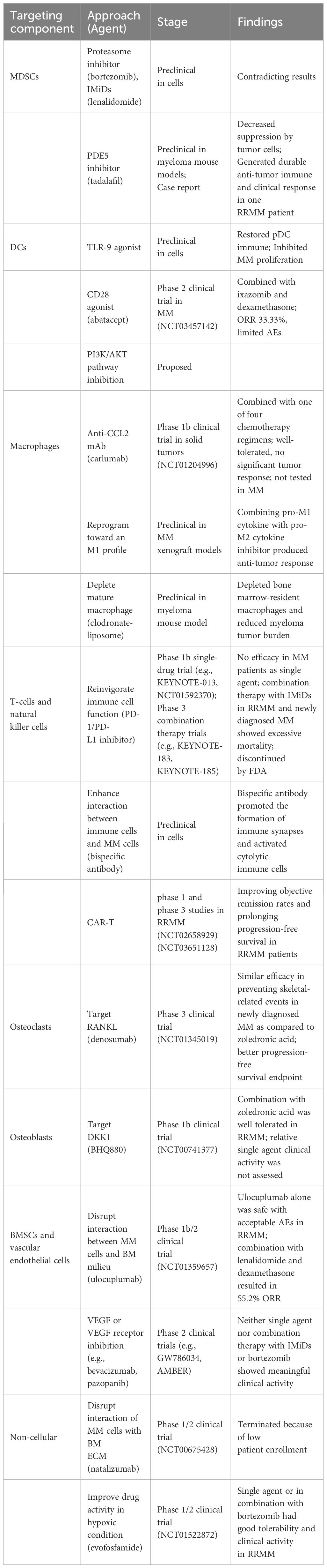
Table 1 Strategies targeting BM microenvironment of MM.
Given that currently available medicines like the proteasome inhibitor and the immunomodulatory drug target both myeloma cells and the MM milieu, the influence of these treatments on MDSCs has been investigated. Görgün and colleagues showed neither bortezomib nor lenalidomide eliminated the amount or suppressive activity of MDSCs in vitro (138). However, in contrast, another study from Wang et al. discovered a decrease in MDSCs after proteasome inhibitor therapy (139). These contradicting results indicate that the impact of currently accessible treatments on MM MDSCs is still debatable and requires further research. Phosphodiesterase-5 (PDE5) inhibition in preclinical studies has demonstrated down-regulation of expression of arginase 1 and nitric oxide synthase-2 in murine tumor models, thereby decreasing the suppressive machinery of MDSCs recruited by tumor cells (140). Noonan et al. reported a case where the addition of tadalafil, a PDE5 inhibitor, restored the responsiveness to lenalidomide-based therapy in one patient who was previously refractory to lenalidomide (141). These data indicate that strategies targeting the function and amount of MDSCs with PDE5 inhibitors may provide a unique strategy that can synergize with tumor-directed therapies to generate a significant and long-lasting anti-myeloma immune and clinical response.
Currently explored MM therapeutic strategies involve pDC immune function restoration and DCs-mediated myeloma proliferation inhibition. Preclinical data have demonstrated that Toll-like receptor 9 (TLR-9) agonists can restore pDC immune function and simultaneously abrogate pDC-induced MM cell growth (142). Unfortunately, there is no clinical trial of TLR-9 agonist in MM patients so far. In vitro studies have demonstrated that blocking of RANKL: APRIL and CD28: CD80/CD86 pathways can suppress myeloma growth mediated by DCs and resensitize tumor plasma cells to lysis by cytotoxic T cells (21, 22). Currently, a synthetic antagonist of CD28, abatacept is being assessed in conjunction with ixazomib and dexamethasone in a phase 2 clinical trial (NCT03457142) for participants with chemotherapy-resistant multiple myeloma. Another therapeutic target is suggested by Megan et al. as they reported that CD28 pro-survival signaling relies on the phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) pathway (143). This pathway is aberrantly activated through numerous mechanisms in most MM patients and is essential for MM survival and chemotherapy resistance (23). As such, PI3K/AKT pathway inhibitors may block CD28 signaling and resensitize MM cells to chemotherapies.
Existing therapeutic approaches targeting macrophages mainly include blocking their recruitment to the tumor microenvironment, reprogramming TAMs, inhibiting the CD47/SIRPα checkpoint, and reversing drug resistance (35). Clodronate Liposome directly deplete BM-resident macrophages and disrupts MM cell homing (144). Reducing monocyte/macrophage recruitment by modulating growth factors, chemokines and cytokines in tumor and stromal cells also diminishes the supportive role of TAMs. The CXCL12-CXCR4 signaling pathway is crucial for macrophage recruitment (145). High expression of the chemokine CXCL12 in MM cells promotes monocyte recruitment and differentiation into an M2 phenotype, which enhances angiogenesis and immunosuppression (29). CXCR4 antibody significantly reduces monocyte recruitment. MM secretes CCL2, which induces the expression of monocyte chemotactic protein-1-induced protein 1(MCPIP1) in macrophages via the JAK2-STAT3 pathway, promoting macrophage homing, proliferation, and M2-like phenotypic polarization (33, 34). Consequently, CCR2 monoclonal antibodies or inhibitors can disrupt macrophage recruitment in MM (146, 147).
Reprogramming TAMs involves decreasing the M2 immunosuppressive phenotype and increasing the M1 phenotype. Gutiérrez-González et al. suggest that the combination of pro-M1 cytokine granulocyte-macrophage CSF (GM-CSF) and pro-M2 cytokine macrophage migration inhibitory factor inhibitor has significant anti-tumor effects (148). Preclinical studies have shown that the JAK1/2 inhibitor Ruxolitinib reduces the M2 polarized phenotype and increases the M1 polarized phenotype by downregulating Tribbles Homolog 1 protein kinase expression (149). Both CD40 agonists and blockers of IL-10R have been shown to reprogram TAMs (150, 151).
Regarding immune checkpoints, numerous CD47-targeted drugs, including anti-CD47 monoclonal antibodies and SIPRα fusion proteins, are under clinical investigation (152). AO-176 is a humanized IgG2 anti-cd47 monoclonal antibody, is being evaluated in a phase 1/2 clinical study to assess its efficacy as a monotherapy and in combination with bortezomib/dexamethasone for the treatment of MM (NCT04445701).
Reinvigorating T-cells and NK cells is considered a compelling and promising therapeutic option in MM. As previously discussed, the increased expression of immune checkpoints in T- and NK cells in MM results in impaired cytolytic immune cell function and the establishment of a tumor-promoting and immune-suppressive microenvironment. Inhibition of PD-1/PD-L1 has shown potential as a treatment for myeloma based on preclinical evidence (153, 154). However, early-phase clinical trials targeting PD-1/PD-L1 showed no efficacy when using a single-drug approach (155, 156). As a result, subsequent studies have focused on investigating the blockade of the PD-1 axis as part of a combination treatment approach with immunomodulatory drugs (IMiDs). One of the IMiDs, lenalidomide, has been proven to enhance checkpoint blockade-induced MM cytotoxicity in a preclinical study (154). Encouragingly, early clinical studies have shown acceptable safety and durable responses of this treatment (157). Unfortunately, two phase 3 trials (KEYNOTE-183 and KEYNOTE-185) evaluating the efficacy and safety of combining PD-1 inhibitor pembrolizumab with IMiDs for the treatment of RRMM and newly diagnosed MM revealed excessive mortality (158, 159). These adverse outcomes led to the discontinuation of these two clinical trials by the Food and Drug Administration (FDA), along with several other similar studies. Based on these findings, the key points in future studies should be to identify patients who would benefit most from checkpoint targeting, determine appropriate drug combinations, and effectively manage adverse events. Furthermore, ongoing research is exploring the benefit-risk profile of the other immune checkpoint or agonist proteins, such as T-cell immunoreceptor with Ig and ITIM domains (TIGIT), lymphocyte activation gene-3 (LAG3), OX40, and immunoglobulin-like receptors (KIRs), as prospective therapeutic targets for MM, either alone or in combination with MM targeted and immunotherapies (160, 161).
An alternative approach is to enhance the interaction between immune cells and MM cells. The vast majority of cytolytic immune cells, such as CD8+ T cells, NK cells, and NK T cells, express an activation receptor called NKG2D. Meanwhile, CS1 (SLAMF7), a surface lymphocytic activation molecule, is highly expressed on MM cells compared to NK cells and a subset of activated T-cells. Building upon this knowledge, Wing Keung and colleagues engineered a bispecific antibody (biAb) to bring together immune cells and MM cells by combining an anti-CS1 single-chain variable fragment (scFv) with an anti-NKG2D scFv (CS1-NKG2D biAb). They demonstrated that the CS1-NKG2D biAb effectively engaged human MM cell lines and NKG2D+ cytolytic innate and antigen-specific effector cells. This interaction facilitated the formation of immune synapses and activated these immune cells against MM (162). The compelling results from this study warrant further research into the effects and potential benefits of CS1-NKG2D biAb in the context of MM treatment.
Currently, novel T-cell-based immunotherapies such as chimeric antigen receptor (CAR-T) therapy and bispecific T cell engagers (BiTE) have significantly improved the treatment of MM. CAR-T therapy involves genetically modifying a patient’s T cells in vitro to express receptors that target specific antigens on tumor cells, enabling these T cells to effectively eliminate the tumor cells upon transfusion back into the patient. The primary targets for CAR-T in MM include B cell maturation antigen (BCMA), CD138, CD19, and CD38. BCMA is exclusively expressed on the surface of mature B cells, and its overexpression and activation are associated with MM in preclinical models and clinical studies, underscoring its potential as a therapeutic target (163). bb2121(ide-cel), a CAR-T therapy targeting BCMA, has demonstrated promising efficacy in phase I and phase III studies for patients with relapsed or refractory multiple myeloma (NCT02658929) (NCT03651128) (164, 165).
CD38, a transmembrane glycoprotein involved in calcium ion regulation, signal transduction and cell adhesion is highly expressed in B precursor cells, plasma cells, natural killer cells and bone marrow precursor cells. CAR-T therapies co-targeting CD38 and other tumor surface antigens, such as BCMA, have also been employed to treat recurrent MM.
The inflammatory response induced by CAR-T cells can enhance the recognition of neoantigens by the host immune system and trigger the anti-tumor response of the natural immune system. It promoted the recognition of other tumor antigens by unconventional T cells through TCR and helped to target tumor cells that were negative for CAR T cell antigens (166).
BiTE antibody molecules consist of single-chain fragment variable (scFvs) from two monoclonal antibodies that recognize antigens on the surface of target cells and CD3 molecules on the surface of T cells. By binding to both the target cell surface antigen and T cell CD3, BiTEs can activate the proliferation of polyclonal cytotoxic T cells, thereby exerting cytotoxic effects and killing target cells (163).
BiTEs targeting BCMA, GPRC5D (G protein-coupled receptor family C group 5 member D), and FcRH5 (Fc receptor-homologue 5) have shown good efficacy and manageable safety profiles in patients with relapsed/refractory multiple myeloma (RRMM) (167). Multiple clinical studies suggest that bispecific antibodies (BsAbs) provide significant survival benefits for RRMM patients. Currently, there are three BiTEs approved by the FDA for the treatment of multiple myeloma, targeting BCMA and GPRC5D.
Teclistamab, a BCMA × CD3 bispecific antibody, was the first approved for the treatment of RRMM. In the phase 1/2 MajesTEC-1 study, teclistamab demonstrated an overall response rate (ORR) of 63.0% in 165 heavily pretreated patients with RRMM (168, 169). Elranatamab, another BCMA × CD3 bispecific antibody, is used for the treatment of multiple myeloma. The approval of this drug is based on several pivotal clinical trials where elranatamab demonstrated high rates of deep and durable responses, including in patients achieving ≥CR. It also exhibited manageable safety (170, 171).
GPRC5D is a G protein-coupled orphan receptor with high expression on malignant plasma cells and low levels on B cells and bone marrow precursor cells, making it a promising target for RRMM patients. Talquetamab, a first-in-class GPRC5D × CD3 bispecific antibody, showed promising results in the MonumenTAL-1 study (NCT03399799) and (NCT04634552). With a median follow-up of 14.9 months, 8.6 months, and 11.8 months in cohorts receiving 0.4 mg/kg QW, 0.8 mg/kg Q2W, and prior T-cell redirected therapies, talquetamab achieved ORRs of 74%, 73%, and 63%, respectively. Cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS) were observed in 79%, 75%, 77% and 11%, 11%, 3% of patients, respectively. The incidence of grade 3/4 infections was 22%, 16%, and 26%, respectively (172).
FcRH5 is expressed on almost all myeloma cells, with significant higher expression than normal B cells. The FcRH5-targeting BiTEs, Cevostamab (BFCR4350A, RG6160), demonstrated promising results in the GO39775 trial (NCT03275103), achieving an ORR of approximately 52% in RRMM patients, with only one patient (2%) experiencing grade 3 cytokine release syndrome (173).
Tri-specific antibodies, which can simultaneously target two tumor antigens, such as BCMA+GPRC5D/CD3, are under clinical investigation.
However, antibody-based drugs face challenges, particularly related to infection risks, necessitating careful management to prevent and treat infections like Pneumocystis pneumonia and viral infections. Immune-related adverse reactions include cytokine storms and immune-mediated lung injury. With GPRC5D bispecific antibodies, there are concerns about potential central nervous system damage and keratin-related changes (skin and nail alterations, taste loss, dry mouth, and swallowing difficulties). Patients still face significant hurdles and require additional treatment post-relapse (174, 175).
As for OCs, the current focus of MM treatment is to target the protein RANKL, which is overproduced by myeloma cells, bone marrow stromal cells, and osteocytes in MM. This excessive RANKL production leads to increased osteoclast activity which in turn recruits myeloma cells and promotes their proliferation, survival, and resistance to apoptosis (176–179). Denosumab, a human monoclonal antibody with high affinity and specificity for RANKL, has been studied as a potential treatment option. A phase 3 clinical trial conducted by Noopur et al. compared denosumab to zoledronic acid as a treatment for bone disease in newly diagnosed patients with MM. This study showed that denosumab was non-inferior to zoledronic acid in preventing skeletal-related events. Remarkably, denosumab demonstrated superior results in terms of the progression-free survival endpoint, providing further support for the efficacy of anti-RANKL therapy against myeloma. The reduction in risk of renal adverse events further supports the potential of denosumab as an additional treatment option for MM patients (180).
In OBs, their dysfunction is significantly influenced by the Dickkopf-1 (DKK1) produced by MM cells. Preclinical data demonstrated that BHQ880, a human DKK1 neutralizing antibody, could promote osteoblast differentiation while inhibiting the growth of myeloma cell and the formation of osteolytic lesions. These conclusions justify the need for clinical evaluation of BHQ880 in MM patients (181). While BHQ880 was well tolerated in a phase 1b trial showed that, its relative clinical activity could not be confirmed due to the accompanied administration of zoledronic acid and anti-myeloma therapy in the study (182).
Certain MM treatments aim to disrupt the interaction between MM cell and BMSCs and vascular endothelial cells. As previously mentioned, CXCR4 is essential for myeloma cell dissemination within and outside the BM. It acts as a pivotal regulator of EMD formation by inducing an EMT-like phenotype in MM, highlighting CXCR4 as a unique therapeutic target for patients with EMD and late-stage RRMM. Both in vitro and in vivo studies have shown that the downregulation of CXCR4 by therapeutic targeting or knockdown can impair the interaction between MM cells and BM milieu and increase their sensitivity to other therapeutic agents (183), providing a rationale for investigating its usage in clinical settings. Additionally, CXCR4 blockade decreases CD4+ T-cell exhaustion (184), enhances cytotoxic activity of immune cells (185), reverts the suppressive activity of Tregs (186), and modulates immunotherapy with anti-PD-1 (187), emphasizing its immunomodulatory effects. In a phase 1b/2 trial, ulocuplumab, a first-in-class fully human IgG4 monoclonal anti-CXCR4 antibody, demonstrated satisfactory safety and achieved an excellent overall response rate of 55.2% when combined with lenalidomide and dexamethasone in RRMM patients (188). This clinical trial supported CXCR4 inhibitors as an attractive class of anti-myeloma medications that deserve additional evaluation in larger clinical studies. Further research is required to explore the synergy between CXCR4 inhibitors and other anti-MM agents.
Also, the VEGF overexpression resulting from this interaction represents another potential target. Preclinical researches have shown that VEGF inhibition has activity against MM cells and synergizes with the proteasome inhibitor bortezomib (189, 190). However, in several clinical trials testing VEGF or VEGF receptor (VEGFR) inhibitors as single-agent treatment, none of them have shown significant clinical responses (191, 192). Similarly, combination regimens involving these inhibitors with IMiDs or with bortezomib have not yielded meaningful results (192, 193). Hence, future attempts to inhibit VEGF in MM patients ought to be made cautiously in patient selection and supported by a solid justification.
Direct inhibition of VLA-4 has shown numerous beneficial effects in the MM microenvironment in preclinical studies. It can diminish MM stimulation by BM stroma, attenuate the pro-cancer signals initiated by MM-mesenchymal stromal cells (MSCs) microvesicles (MVs), abrogate angiogenesis induced by VEGF, block signaling pathways triggered by VEGF and IGF-1that promote MM cell migration, and restore sensitivity of MM cells treated with MM-MSCs MVs to doxorubicin and bortezomib (194, 195). According to these advantages, natalizumab, a recombinant humanized monoclonal antibody (mAb) against VLA-4 that is approved for Crohn’s disease and multiple sclerosis treatment, was evaluated in a phase 1/2 study (NCT00675428) for RRMM. Regrettably, this trial was prematurely terminated not due to safety concerns but due to insufficient patient enrollment. Therefore, alternative approaches are required to specifically target VLA-4 in the MM microenvironment.
Since hypoxic conditions have been identified in the BM of MM patients, evofosfamide, a 2-nitroimidazole prodrug of the DNA alkylator bromo-isophosphoramide (Br-IPM), has been specifically engineered to exhibit activity under low oxygen conditions, and demonstrated efficacy in both in vitro and in vivo preclinical models of MM (196). Evofosfamide also showed a synergistic effect when combined with bortezomib, leading to the promotion of apoptosis in MM cells (137). A phase 1/2 trial reveals that the administration of evofosfamide, with or without bortezomib, has excellent tolerability and results in stable disease and improved survival in patients with heavy prior treatments with advanced-stage RRMM (197). These data indicate that targeting the hypoxic microenvironment could be an effective new approach to the treatment of MM, emphasizing the need for further investigation.
The intricate interplay between the bone marrow (BM) microenvironment and myeloma cells plays a crucial role in supporting MM cell growth and facilitating immune evasion through diverse mechanisms. As a result, the tumor microenvironment is a promising therapeutic target, particularly for heavily pre-treated patients with end-stage RRMM. As such, multiple therapeutic strategies targeting different MM microenvironment components have been discovered and developed over the past decades to improve patient outcomes. Despite the significant advancements, most of the strategies failed to translate into clinical practice. Therefore, future research endeavors should focus on conducting larger-scale and multicenter studies to elucidate the mechanisms of action and resistance associated with different therapeutic approaches, identify new targets, and develop more effective, selective, and well-tolerated targeted therapies. This will contribute to improving patient outcomes and advancing the field of MM treatment.
KL: Conceptualization, Writing – original draft. WW: Writing – review & editing, Data curation. YL: Writing – original draft. CX: Writing – review & editing. JL: Writing – review & editing. LX: Conceptualization, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grant ZR2021MH072 to LX from Natural Science Foundation of Shandong Province, grant 82200224 to LX from National Natural Science Foundation of China, grant 2023M732124 to LX from China Postdoctoral Science Foundation, and grant 2023HWYQ-115 to LX from Shandong Excellent Youth Science Fund Project (overseas), and grant tsqn202312361to LX from Taishan Scholars Program of Shandong Province.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Rajkumar SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G, Mateos M-V, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. (2014) 15:e538–e48. doi: 10.1016/S1470-2045(14)70442-5
2. Kawano Y, Moschetta M, Manier S, Glavey S, Gorgun GT, Roccaro AM, et al. Targeting the bone marrow microenvironment in multiple myeloma. Immunol Rev. (2015) 263:160–72. doi: 10.1111/imr.12233
3. Hinshaw DC, Shevde LA. The tumor microenvironment innately modulates cancer progression. Cancer Res. (2019) 79:4557–66. doi: 10.1158/0008-5472.CAN-18-3962
4. Garcia-Ortiz A, Rodriguez-Garcia Y, Encinas J, Maroto-Martin E, Castellano E, Teixido J, et al. The role of tumor microenvironment in multiple myeloma development and progression. Cancers (Basel). (2021) 13(2):217. doi: 10.3390/cancers13020217
5. Groth C, Hu X, Weber R, Fleming V, Altevogt P, Utikal J, et al. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br J Cancer. (2019) 120:16–25. doi: 10.1038/s41416-018-0333-1
6. Giannotta C, Autino F, Massaia M. The immune suppressive tumor microenvironment in multiple myeloma: the contribution of myeloid-derived suppressor cells. Front Immunol. (2022) 13:1102471. doi: 10.3389/fimmu.2022.1102471
7. Sui H, Dongye S, Liu X, Xu X, Wang L, Jin CQ, et al. Immunotherapy of targeting MDSCs in tumor microenvironment. Front Immunol. (2022) 13:990463. doi: 10.3389/fimmu.2022.990463
8. Zhou J, Shen Q, Lin H, Hu L, Li G, Zhang X. Decitabine shows potent anti-myeloma activity by depleting monocytic myeloid-derived suppressor cells in the myeloma microenvironment. J Cancer Res Clin Oncol. (2019) 145:329–36. doi: 10.1007/s00432-018-2790-6
9. Smith LK, Boukhaled GM, Condotta SA, Mazouz S, Guthmiller JJ, Vijay R, et al. Interleukin-10 directly inhibits CD8(+) T cell function by enhancing N-glycan branching to decrease antigen sensitivity. Immunity. (2018) 48:299–312.e5. doi: 10.1016/j.immuni.2018.01.006
10. Vladimirovna IL, Sosunova E, Nikolaev A, Nenasheva T. Mesenchymal stem cells and myeloid derived suppressor cells: common traits in immune regulation. J Immunol Res. (2016) 2016:7121580. doi: 10.1155/2016/7121580
11. Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med. (2007) 13:828–35. doi: 10.1038/nm1609
12. Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol. (2021) 21:485–98. doi: 10.1038/s41577-020-00490-y
13. Yu J, Wang Y, Yan F, Zhang P, Li H, Zhao H, et al. Noncanonical NF-κB activation mediates STAT3-stimulated IDO upregulation in myeloid-derived suppressor cells in breast cancer. J Immunol (Baltimore Md: 1950). (2014) 193:2574–86. doi: 10.4049/jimmunol.1400833
14. De Veirman K, Menu E, Maes K, De Beule N, De Smedt E, Maes A, et al. Myeloid-derived suppressor cells induce multiple myeloma cell survival by activating the AMPK pathway. Cancer Lett. (2019) 442:233–41. doi: 10.1016/j.canlet.2018.11.002
15. Ai L, Mu S, Sun C, Fan F, Yan H, Qin Y, et al. Myeloid-derived suppressor cells endow stem-like qualities to multiple myeloma cells by inducing piRNA-823 expression and DNMT3B activation. Mol Cancer. (2019) 18:88. doi: 10.1186/s12943-019-1011-5
16. Nakamura K, Kassem S, Cleynen A, Chretien ML, Guillerey C, Putz EM, et al. Dysregulated IL-18 is a key driver of immunosuppression and a possible therapeutic target in the multiple myeloma microenvironment. Cancer Cell. (2018) 33:634–48 e5. doi: 10.1016/j.ccell.2018.02.007
17. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. (2013) 31:563–604. doi: 10.1146/annurev-immunol-020711-074950
18. Wang S, Hong S, Yang J, Qian J, Zhang X, Shpall E, et al. Optimizing immunotherapy in multiple myeloma: Restoring the function of patients' monocyte-derived dendritic cells by inhibiting p38 or activating MEK/ERK MAPK and neutralizing interleukin-6 in progenitor cells. Blood. (2006) 108:4071–7. doi: 10.1182/blood-2006-04-016980
19. Brimnes MK, Svane IM, Johnsen HE. Impaired functionality and phenotypic profile of dendritic cells from patients with multiple myeloma. Clin Exp Immunol. (2006) 144:76–84. doi: 10.1111/j.1365-2249.2006.03037.x
20. Shinde P, Fernandes S, Melinkeri S, Kale V, Limaye L. Compromised functionality of monocyte-derived dendritic cells in multiple myeloma patients may limit their use in cancer immunotherapy. Sci Rep. (2018) 8:5705. doi: 10.1038/s41598-018-23943-w
21. Kukreja A, Hutchinson A, Dhodapkar K, Mazumder A, Vesole D, Angitapalli R, et al. Enhancement of clonogenicity of human multiple myeloma by dendritic cells. J Exp Med. (2006) 203:1859–65. doi: 10.1084/jem.20052136
22. Leone P, Berardi S, Frassanito MA, Ria R, De Re V, Cicco S, et al. Dendritic cells accumulate in the bone marrow of myeloma patients where they protect tumor plasma cells from CD8+ T-cell killing. Blood. (2015) 126:1443–51. doi: 10.1182/blood-2015-01-623975
23. Murray ME, Gavile CM, Nair JR, Koorella C, Carlson LM, Buac D, et al. CD28-mediated pro-survival signaling induces chemotherapeutic resistance in multiple myeloma. Blood. (2014) 123:3770–9. doi: 10.1182/blood-2013-10-530964
24. Chauhan D, Singh AV, Brahmandam M, Carrasco R, Bandi M, Hideshima T, et al. Functional interaction of plasmacytoid dendritic cells with multiple myeloma cells: a therapeutic target. Cancer Cell. (2009) 16:309–23. doi: 10.1016/j.ccr.2009.08.019
25. Ray A, Buon L, Song Y, Chauhan D, Anderson KC. Interaction of plasmacytoid dendritic cells and myeloma cells trigger tumor promoting transcriptional changes in multiple myeloma cells. Blood. (2019) 134:510. doi: 10.1182/blood-2019-123333
26. Pan Y, Yu Y, Wang X, Zhang T. Tumor-associated macrophages in tumor immunity. Front Immunol. (2020) 11:583084. doi: 10.3389/fimmu.2020.583084
27. Petty AJ, Yang Y. Tumor-associated macrophages in hematologic Malignancies: new insights and targeted therapies. Cells. (2019) 8(12):1526. doi: 10.3390/cells8121526
28. Ribatti D, Moschetta M, Vacca A. Macrophages in multiple myeloma. Immunol Lett. (2014) 161:241–4. doi: 10.1016/j.imlet.2013.12.010
29. Beider K, Bitner H, Leiba M, Gutwein O, Koren-Michowitz M, Ostrovsky O, et al. Multiple myeloma cells recruit tumor-supportive macrophages through the CXCR4/CXCL12 axis and promote their polarization toward the M2 phenotype. Oncotarget. (2014) 5:11283–96. doi: 10.18632/oncotarget.v5i22
30. Wang SSY, Chng WJ, Liu H, de Mel S. Tumor-associated macrophages and related myelomonocytic cells in the tumor microenvironment of multiple myeloma. Cancers (Basel). (2022) 14(22):5654. doi: 10.3390/cancers14225654
31. Li J, Yang Y, Wang W, Xu J, Sun Y, Jiang J, et al. Single-cell atlas of the immune microenvironment reveals macrophage reprogramming and the potential dual macrophage-targeted strategy in multiple myeloma. Br J hHaematol. (2023) 201:917–34. doi: 10.1111/bjh.18708
32. Gutierrez-Gonzalez A, Martinez-Moreno M, Samaniego R, Arellano-Sanchez N, Salinas-Munoz L, Relloso M, et al. Evaluationxxx of xxxthe xxxpotential xxxtherapeutic benefits of macrophage reprogramming in multiple myeloma. Blood. (2016) 128:2241–52. doi: 10.1182/blood-2016-01-695395
33. Li Y, Zheng Y, Li T, Wang Q, Qian J, Lu Y, et al. Chemokines CCL2, 3, 14 stimulate macrophage bone marrow homing, proliferation, and polarization in multiple myeloma. Oncotarget. (2015) 6:24218–29. doi: 10.18632/oncotarget.v6i27
34. Xu R, Li Y, Yan H, Zhang E, Huang X, Chen Q, et al. CCL2 promotes macrophages-associated chemoresistance via MCPIP1 dual catalytic activities in multiple myeloma. Cell Death Dis. (2019) 10:781. doi: 10.1038/s41419-019-2012-4
35. Sun J, Park C, Guenthner N, Gurley S, Zhang L, Lubben B, et al. Tumor-associated macrophages in multiple myeloma: advances in biology and therapy. J Immunother Cancer. (2022) 10(4):e003975. doi: 10.1136/jitc-2021-003975
36. Kim J, Denu RA, Dollar BA, Escalante LE, Kuether JP, Callander NS, et al. Macrophages and mesenchymal stromal cells support survival and proliferation of multiple myeloma cells. Br J Haematol. (2012) 158:336–46. doi: 10.1111/j.1365-2141.2012.09154.x
37. Gadó K, Domján G, Hegyesi H, Falus A. Role of INTERLEUKIN-6 in the pathogenesis of multiple myeloma. Cell Biol Int. (2000) 24:195–209. doi: 10.1006/cbir.2000.0497
38. Kovacs E. Interleukin-6 leads to interleukin-10 production in several human multiple myeloma cell lines. Does interleukin-10 enhance the proliferation of these cells? Leukemia Res. (2010) 34:912–6. doi: 10.1016/j.leukres.2009.08.012
39. Akhmetzyanova I, Aaron T, Galbo P, Tikhonova A, Dolgalev I, Tanaka M, et al. Tissue-resident macrophages promote early dissemination of multiple myeloma via IL-6 and TNFalpha. Blood Adv. (2021) 5:3592–608. doi: 10.1182/bloodadvances.2021005327
40. Ullah TR. The role of CXCR4 in multiple myeloma: Cells' journey from bone marrow to beyond. J Bone Oncol. (2019) 17:100253. doi: 10.1016/j.jbo.2019.100253
41. Sun J, Muz B, Alhallak K, Markovic M, Gurley S, Wang Z, et al. Targeting CD47 as a novel immunotherapy for multiple myeloma. Cancers (Basel). (2020) 12(2):305. doi: 10.3390/cancers12020305
42. Scavelli C, Nico B, Cirulli T, Ria R, Di Pietro G, Mangieri D, et al. Vasculogenic mimicry by bone marrow macrophages in patients with multiple myeloma. Oncogene. (2008) 27:663–74. doi: 10.1038/sj.onc.1210691
43. Aras S, Zaidi MR. TAMeless traitors: macrophages in cancer progression and metastasis. Br J cancer. (2017) 117:1583–91. doi: 10.1038/bjc.2017.356
44. Sun M, Qiu S, Xiao Q, Wang T, Tian X, Chen C, et al. Synergistic effects of multiple myeloma cells and tumor-associated macrophages on vascular endothelial cells in vitro. Med Oncol (Northwood London England). (2020) 37:99. doi: 10.1007/s12032-020-01426-1
45. Rastgoo N, Wu J, Liu A, Pourabdollah M, Atenafu EG, Reece D, et al. Targeting CD47/TNFAIP8 by miR-155 overcomes drug resistance and inhibits tumor growth through induction of phagocytosis and apoptosis in multiple myeloma. Haematologica. (2020) 105:2813–23. doi: 10.3324/haematol.2019.227579
46. Alexandrakis MG, Goulidaki N, Pappa CA, Boula A, Psarakis F, Neonakis I, et al. Interleukin-10 induces both plasma cell proliferation and angiogenesis in multiple myeloma. Pathol Oncol Res: POR. (2015) 21:929–34. doi: 10.1007/s12253-015-9921-z
47. Camiolo G, Barbato A, Giallongo C, Vicario N, Romano A, Parrinello NL, et al. Iron regulates myeloma cell/macrophage interaction and drives resistance to bortezomib. Redox Biol. (2020) 36:101611. doi: 10.1016/j.redox.2020.101611
48. De Beule N, De Veirman K, Maes K, De Bruyne E, Menu E, Breckpot K, et al. Tumour-associated macrophage-mediated survival of myeloma cells through STAT3 activation. J Pathol. (2017) 241:534–46. doi: 10.1002/path.4860
49. Beyar-Katz O, Magidey K, Reiner-Benaim A, Barak N, Avivi I, Cohen Y, et al. Proinflammatory macrophages promote multiple myeloma resistance to bortezomib therapy. Mol Cancer Res: MCR. (2019) 17:2331–40. doi: 10.1158/1541-7786.MCR-19-0487
50. Chen J, He D, Chen Q, Guo X, Yang L, Lin X, et al. BAFF is involved in macrophage-induced bortezomib resistance in myeloma. Cell Death Dis. (2017) 8:e3161. doi: 10.1038/cddis.2017.533
51. Yan ZX, Li L, Wang W, OuYang BS, Cheng S, Wang L, et al. Clinical efficacy and tumor microenvironment influence in a dose-escalation study of anti-CD19 chimeric antigen receptor T cells in refractory B-cell non-hodgkin's lymphoma. Clin Cancer research: an Off J Am Assoc Cancer Res. (2019) 25:6995–7003. doi: 10.1158/1078-0432.CCR-19-0101
52. Golstein P, Griffiths GM. An early history of T cell-mediated cytotoxicity. Nat Rev Immunol. (2018) 18:527–35. doi: 10.1038/s41577-018-0009-3
53. Bailur JK, McCachren SS, Doxie DB, Shrestha M, Pendleton K, Nooka AK, et al. Early alterations in stem-like/resident T cells, innate and myeloid cells in the bone marrow in preneoplastic gammopathy. JCI Insight. (2019) 5(11):e127807. doi: 10.1172/jci.insight.127807
54. Paiva B, Mateos MV, Sanchez-Abarca LI, Puig N, Vidriales MB, Lopez-Corral L, et al. Immune status of high-risk smoldering multiple myeloma patients and its therapeutic modulation under LenDex: a longitudinal analysis. Blood. (2016) 127:1151–62. doi: 10.1182/blood-2015-10-662320
55. Ray A, Das DS, Song Y, Richardson P, Munshi NC, Chauhan D, et al. Targeting PD1-PDL1 immune checkpoint in plasmacytoid dendritic cell interactions with T cells, natural killer cells and multiple myeloma cells. Leukemia. (2015) 29:1441–4. doi: 10.1038/leu.2015.11
56. Atanackovic D, Luetkens T, Kroger N. Coinhibitory molecule PD-1 as a potential target for the immunotherapy of multiple myeloma. Leukemia. (2014) 28:993–1000. doi: 10.1038/leu.2013.310
57. Guillerey C, Harjunpaa H, Carrie N, Kassem S, Teo T, Miles K, et al. TIGIT immune checkpoint blockade restores CD8(+) T-cell immunity against multiple myeloma. Blood. (2018) 132:1689–94. doi: 10.1182/blood-2018-01-825265
58. Dhodapkar KM. Role of tissue-resident memory in intra-tumor heterogeneity and response to immune checkpoint blockade. Front Immunol. (2018) 9:1655. doi: 10.3389/fimmu.2018.01655
59. Saravia J, Chapman NM, Chi H. Helper T cell differentiation. Cell Mol Immunol. (2019) 16:634–43. doi: 10.1038/s41423-019-0220-6
60. Haabeth OA, Lorvik KB, Hammarstrom C, Donaldson IM, Haraldsen G, Bogen B, et al. Inflammation driven by tumour-specific Th1 cells protects against B-cell cancer. Nat Commun. (2011) 2:240. doi: 10.1038/ncomms1239
61. Feng X, Zhang L, Acharya C, An G, Wen K, Qiu L, et al. Targeting CD38 suppresses induction and function of T regulatory cells to mitigate immunosuppression in multiple myeloma. Clin Cancer Res. (2017) 23:4290–300. doi: 10.1158/1078-0432.CCR-16-3192
62. Brown R, Kabani K, Favaloro J, Yang S, Ho PJ, Gibson J, et al. CD86+ or HLA-G+ can be transferred via trogocytosis from myeloma cells to T cells and are associated with poor prognosis. Blood. (2012) 120:2055–63. doi: 10.1182/blood-2012-03-416792
63. Tai YT, Lin L, Xing L, Cho SF, Yu T, Acharya C, et al. APRIL signaling via TACI mediates immunosuppression by T regulatory cells in multiple myeloma: therapeutic implications. Leukemia. (2019) 33:426–38. doi: 10.1038/s41375-018-0242-6
64. Kawano Y, Zavidij O, Park J, Moschetta M, Kokubun K, Mouhieddine TH, et al. Blocking IFNAR1 inhibits multiple myeloma-driven Treg expansion and immunosuppression. J Clin Invest. (2018) 128:2487–99. doi: 10.1172/JCI88169
65. Frassanito MA, Ruggieri S, Desantis V, Di Marzo L, Leone P, Racanelli V, et al. Myeloma cells act as tolerogenic antigen-presenting cells and induce regulatory T cells in vitro. Eur J Haematol. (2015) 95:65–74. doi: 10.1111/ejh.12481
66. Alrasheed N, Lee L, Ghorani E, Henry JY, Conde L, Chin M, et al. Marrow-infiltrating regulatory T cells correlate with the presence of dysfunctional CD4(+)PD-1(+) cells and inferior survival in patients with newly diagnosed multiple myeloma. Clin Cancer Res. (2020) 26:3443–54. doi: 10.1158/1078-0432.CCR-19-1714
67. Wang L, Yi T, Kortylewski M, Pardoll DM, Zeng D, Yu H. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med. (2009) 206:1457–64. doi: 10.1084/jem.20090207
68. Alexandrakis MG, Pappa CA, Miyakis S, Sfiridaki A, Kafousi M, Alegakis A, et al. Serum interleukin-17 and its relationship to angiogenic factors in multiple myeloma. Eur J Intern Med. (2006) 17:412–6. doi: 10.1016/j.ejim.2006.02.012
69. Prabhala RH, Pelluru D, Fulciniti M, Prabhala HK, Nanjappa P, Song W, et al. Elevated IL-17 produced by TH17 cells promotes myeloma cell growth and inhibits immune function in multiple myeloma. Blood. (2010) 115:5385–92. doi: 10.1182/blood-2009-10-246660
70. Noonan K, Marchionni L, Anderson J, Pardoll D, Roodman GD, Borrello I. A novel role of IL-17-producing lymphocytes in mediating lytic bone disease in multiple myeloma. Blood. (2010) 116:3554–63. doi: 10.1182/blood-2010-05-283895
71. Lorvik KB, Hammarstrom C, Fauskanger M, Haabeth OA, Zangani M, Haraldsen G, et al. Adoptive transfer of tumor-specific th2 cells eradicates tumors by triggering an in situ inflammatory immune response. Cancer Res. (2016) 76:6864–76. doi: 10.1158/0008-5472.CAN-16-1219
72. Szmania S, Lapteva N, Garg T, Greenway A, Lingo J, Nair B, et al. Ex vivo-expanded natural killer cells demonstrate robust proliferation in vivo in high-risk relapsed multiple myeloma patients. J Immunother (Hagerstown Md: 1997). (2015) 38:24–36. doi: 10.1097/CJI.0000000000000059
73. Knight A, Mackinnon S, Lowdell MW. Human Vdelta1 gamma-delta T cells exert potent specific cytotoxicity against primary multiple myeloma cells. Cytotherapy. (2012) 14:1110–8. doi: 10.3109/14653249.2012.700766
74. Gherardin NA, Loh L, Admojo L, Davenport AJ, Richardson K, Rogers A, et al. Enumeration, functional responses and cytotoxic capacity of MAIT cells in newly diagnosed and relapsed multiple myeloma. Sci Rep. (2018) 8:4159. doi: 10.1038/s41598-018-22130-1
75. Godfrey J, Benson DM Jr. The role of natural killer cells in immunity against multiple myeloma. Leuk Lymphoma. (2012) 53:1666–76. doi: 10.3109/10428194.2012.676175
76. Costello RT, Boehrer A, Sanchez C, Mercier D, Baier C, Le Treut T, et al. Differential expression of natural killer cell activating receptors in blood versus bone marrow in patients with monoclonal gammopathy. Immunology. (2013) 139:338–41. doi: 10.1111/imm.12082
77. Jinushi M, Vanneman M, Munshi NC, Tai YT, Prabhala RH, Ritz J, et al. MHC class I chain-related protein A antibodies and shedding are associated with the progression of multiple myeloma. Proc Natl Acad Sci USA. (2008) 105:1285–90. doi: 10.1073/pnas.0711293105
78. Feyler S, Scott GB, Parrish C, Jarmin S, Evans P, Short M, et al. Tumour cell generation of inducible regulatory T-cells in multiple myeloma is contact-dependent and antigen-presenting cell-independent. PloS One. (2012) 7:e35981. doi: 10.1371/journal.pone.0035981
79. Zhang L, Tai YT, Ho M, Xing L, Chauhan D, Gang A, et al. Regulatory B cell-myeloma cell interaction confers immunosuppression and promotes their survival in the bone marrow milieu. Blood Cancer J. (2017) 7:e547. doi: 10.1038/bcj.2017.24
80. Roodman GD. Pathogenesis of myeloma bone disease. Cell Mol Dis. (2004) 32:290–2. doi: 10.1016/j.bcmd.2004.01.001
81. Mansour A, Wakkach A, Blin-Wakkach C. Emerging roles of osteoclasts in the modulation of bone microenvironment and immune suppression in multiple myeloma. Front Immunol. (2017) 8:954. doi: 10.3389/fimmu.2017.00954
82. Abe M, Hiura K, Wilde J, Moriyama K, Hashimoto T, Ozaki S, et al. Role for macrophage inflammatory protein (MIP)-1alpha and MIP-1beta in the development of osteolytic lesions in multiple myeloma. Blood. (2002) 100:2195–202. doi: 10.1182/blood.V100.6.2195
83. Choi SJ, Oba Y, Gazitt Y, Alsina M, Cruz J, Anderson J, et al. Antisense inhibition of macrophage inflammatory protein 1-α blocks bone destruction in a model of myeloma bone disease. J Clin Invest. (2001) 108:1833–41. doi: 10.1172/JCI200113116
84. Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. (2006) 203:2673–82. doi: 10.1084/jem.20061775
85. Sezer O, Heider U, Zavrski I, Kuhne CA, Hofbauer LC. and osteoprotegerin in myeloma bone disease. Blood. (2003) 101:2094–8. doi: 10.1182/blood-2002-09-2684
86. Silbermann R, Bolzoni M, Storti P, Guasco D, Bonomini S, Zhou D, et al. Bone marrow monocyte-/macrophage-derived activin A mediates the osteoclastogenic effect of IL-3 in multiple myeloma. Leukemia. (2014) 28:951–4. doi: 10.1038/leu.2013.385
87. Vallet S, Mukherjee S, Vaghela N, Hideshima T, Fulciniti M, Pozzi S, et al. Activin A promotes multiple myeloma-induced osteolysis and is a promising target for myeloma bone disease. Proc Natl Acad Sci USA. (2010) 107:5124–9. doi: 10.1073/pnas.0911929107
88. Wakkach A, Mansour A, Dacquin R, Coste E, Jurdic P, Carle GF, et al. Bone marrow microenvironment controls the in vivo differentiation of murine dendritic cells into osteoclasts. Blood. (2008) 112:5074–83. doi: 10.1182/blood-2008-01-132787
89. Laperine O, Blin-Wakkach C, Guicheux J, Beck-Cormier S, Lesclous P. Dendritic-cell-derived osteoclasts: a new game changer in bone-resorption-associated diseases. Drug Discov Today. (2016) 21:1345–54. doi: 10.1016/j.drudis.2016.04.022
90. Tucci M, Stucci S, Savonarola A, Ciavarella S, Cafforio P, Dammacco F, et al. Immature dendritic cells in multiple myeloma are prone to osteoclast-like differentiation through interleukin-17A stimulation. Br J Haematol. (2013) 161:821–31. doi: 10.1111/bjh.12333
91. Yaccoby S, Ling W, Zhan F, Walker R, Barlogie B, Shaughnessy JD Jr. Antibody-based inhibition of DKK1 suppresses tumor-induced bone resorption and multiple myeloma growth in vivo. Blood. (2007) 109:2106–11. doi: 10.1182/blood-2006-09-047712
92. Giuliani N, Colla S, Morandi F, Lazzaretti M, Sala R, Bonomini S, et al. Myeloma cells block RUNX2/CBFA1 activity in human bone marrow osteoblast progenitors and inhibit osteoblast formation and differentiation. Blood. (2005) 106:2472–83. doi: 10.1182/blood-2004-12-4986
93. Winkler DG, Sutherland MK, Geoghegan JC, Yu C, Hayes T, Skonier JE, et al. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. (2003) 22:6267–76. doi: 10.1093/emboj/cdg599
94. Baron R, Rawadi G. Wnt signaling and the regulation of bone mass. Curr osteoporosis Rep. (2007) 5:73–80. doi: 10.1007/s11914-007-0006-0
95. Wu M, Chen G, Li YP. TGF-β and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res. (2016) 4:16009. doi: 10.1038/boneres.2016.9
96. An G, Acharya C, Feng X, Wen K, Zhong M, Zhang L, et al. Osteoclasts promote immune suppressive microenvironment in multiple myeloma: therapeutic implication. Blood. (2016) 128:1590–603. doi: 10.1182/blood-2016-03-707547
97. Delgado-Calle J, Anderson J, Cregor MD, Hiasa M, Chirgwin JM, Carlesso N, et al. Bidirectional notch signaling and osteocyte-derived factors in the bone marrow microenvironment promote tumor cell proliferation and bone destruction in multiple myeloma. Cancer Res. (2016) 76:1089–100. doi: 10.1158/0008-5472.CAN-15-1703
98. Chen Z, Orlowski RZ, Wang M, Kwak L, McCarty N. Osteoblastic niche supports the growth of quiescent multiple myeloma cells. Blood. (2014) 123:2204–8. doi: 10.1182/blood-2013-07-517136
99. Lawson MA, McDonald MM, Kovacic N, Hua Khoo W, Terry RL, Down J, et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat Commun. (2015) 6:8983. doi: 10.1038/ncomms9983
100. Cackowski FC, Anderson JL, Patrene KD, Choksi RJ, Shapiro SD, Windle JJ, et al. Osteoclasts are important for bone angiogenesis. Blood. (2010) 115:140–9. doi: 10.1182/blood-2009-08-237628
101. Tanaka Y, Abe M, Hiasa M, Oda A, Amou H, Nakano A, et al. Myeloma cell-osteoclast interaction enhances angiogenesis together with bone resorption: a role for vascular endothelial cell growth factor and osteopontin. Clin Cancer Res. (2007) 13:816–23. doi: 10.1158/1078-0432.CCR-06-2258
102. Khoo WH, Ledergor G, Weiner A, Roden DL, Terry RL, McDonald MM, et al. A niche-dependent myeloid transcriptome signature defines dormant myeloma cells. Blood. (2019) 134:30–43. doi: 10.1182/blood.2018880930
103. de Jong MME, Kellermayer Z, Papazian N, Tahri S, Hofste Op Bruinink D, Hoogenboezem R, et al. The multiple myeloma microenvironment is defined by an inflammatory stromal cell landscape. Nat Immunol. (2021) 22:769–80. doi: 10.1038/s41590-021-00931-3
104. de Jong MME, Fokkema C, Papazian N, Czeti Á, Appelman MK, Vermeulen M, et al. An IL-1β-driven neutrophil-stromal cell axis fosters a BAFF-rich protumor microenvironment in individuals with multiple myeloma. Nat Immunol. (2024) 25:820–33. doi: 10.1038/s41590-024-01808-x
105. Kim CH, Broxmeyer HE. In vitro behavior of hematopoietic progenitor cells under the influence of chemoattractants: stromal cell-derived factor-1, steel factor, and the bone marrow environment. Blood. (1998) 91:100–10. doi: 10.1182/blood.V91.1.100
106. Hideshima T, Chauhan D, Hayashi T, Podar K, Akiyama M, Gupta D, et al. The biological sequelae of stromal cell-derived factor-1alpha in multiple myeloma. Mol Cancer Ther. (2002) 1:539–44.
107. Waldschmidt JM, Simon A, Wider D, Muller SJ, Follo M, Ihorst G, et al. CXCL12 and CXCR7 are relevant targets to reverse cell adhesion-mediated drug resistance in multiple myeloma. Br J Haematol. (2017) 179:36–49. doi: 10.1111/bjh.14807
108. Roccaro AM, Mishima Y, Sacco A, Moschetta M, Tai YT, Shi J, et al. CXCR4 regulates extra-medullary myeloma through epithelial-mesenchymal-transition-like transcriptional activation. Cell Rep. (2015) 12:622–35. doi: 10.1016/j.celrep.2015.06.059
109. Damiano JS, Dalton WS. Integrin-mediated drug resistance in multiple myeloma. Leuk Lymphoma. (2000) 38:71–81. doi: 10.3109/10428190009060320
110. Neri P, Ren L, Azab AK, Brentnall M, Gratton K, Klimowicz AC, et al. Integrin beta7-mediated regulation of multiple myeloma cell adhesion, migration, and invasion. Blood. (2011) 117:6202–13. doi: 10.1182/blood-2010-06-292243
111. Landowski TH, Olashaw NE, Agrawal D, Dalton WS. Cell adhesion-mediated drug resistance (CAM-DR) is associated with activation of NF-kappa B (RelB/p50) in myeloma cells. Oncogene. (2003) 22:2417–21. doi: 10.1038/sj.onc.1206315
112. Hosen N, Matsunaga Y, Hasegawa K, Matsuno H, Nakamura Y, Makita M, et al. The activated conformation of integrin beta7 is a novel multiple myeloma-specific target for CAR T cell therapy. Nat Med. (2017) 23:1436–43. doi: 10.1038/nm.4431
113. Hideshima T, Mitsiades C, Tonon G, Richardson PG, Anderson KC. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat Rev Cancer. (2007) 7:585–98. doi: 10.1038/nrc2189
114. Reijmers RM, Groen RW, Rozemuller H, Kuil A, de Haan-Kramer A, Csikos T, et al. Targeting EXT1 reveals a crucial role for heparan sulfate in the growth of multiple myeloma. Blood. (2010) 115:601–4. doi: 10.1182/blood-2009-02-204396
115. Colombo M, Giannandrea D, Lesma E, Basile A, Chiaramonte R. Extracellular vesicles enhance multiple myeloma metastatic dissemination. Int J Mol Sci. (2019) 20(13):3236. doi: 10.3390/ijms20133236
116. Schillaci O, Fontana S, Monteleone F, Taverna S, Di Bella MA, Di Vizio D, et al. Exosomes from metastatic cancer cells transfer amoeboid phenotype to non-metastatic cells and increase endothelial permeability: their emerging role in tumor heterogeneity. Sci Rep. (2017) 7:4711. doi: 10.1038/s41598-017-05002-y
117. Li B, Hong J, Hong M, Wang Y, Yu T, Zang S, et al. piRNA-823 delivered by multiple myeloma-derived extracellular vesicles promoted tumorigenesis through re-educating endothelial cells in the tumor environment. Oncogene. (2019) 38:5227–38. doi: 10.1038/s41388-019-0788-4
118. Zarfati M, Avivi I, Brenner B, Katz T, Aharon A. Extracellular vesicles of multiple myeloma cells utilize the proteasome inhibitor mechanism to moderate endothelial angiogenesis. Angiogenesis. (2019) 22:185–96. doi: 10.1007/s10456-018-9649-y
119. Raimondi L, De Luca A, Amodio N, Manno M, Raccosta S, Taverna S, et al. Involvement of multiple myeloma cell-derived exosomes in osteoclast differentiation. Oncotarget. (2015) 6:13772–89. doi: 10.18632/oncotarget.v6i15
120. Bandari SK, Purushothaman A, Ramani VC, Brinkley GJ, Chandrashekar DS, Varambally S, et al. Chemotherapy induces secretion of exosomes loaded with heparanase that degrades extracellular matrix and impacts tumor and host cell behavior. Matrix Biol. (2018) 65:104–18. doi: 10.1016/j.matbio.2017.09.001
121. Faict S, Muller J, De Veirman K, De Bruyne E, Maes K, Vrancken L, et al. Exosomes play a role in multiple myeloma bone disease and tumor development by targeting osteoclasts and osteoblasts. Blood Cancer J. (2018) 8:105. doi: 10.1038/s41408-018-0139-7
122. Li B, Xu H, Han H, Song S, Zhang X, Ouyang L, et al. Exosome-mediated transfer of lncRUNX2-AS1 from multiple myeloma cells to MSCs contributes to osteogenesis. Oncogene. (2018) 37:5508–19. doi: 10.1038/s41388-018-0359-0
123. Cheng Q, Li X, Liu J, Ye Q, Chen Y, Tan S, et al. Multiple Myeloma-Derived Exosomes Regulate the Functions of Mesenchymal Stem Cells Partially via Modulating miR-21 and miR-146a. Stem Cells Int. (2017) 2017:9012152. doi: 10.1155/2017/9012152
124. Wang J, De Veirman K, Faict S, Frassanito MA, Ribatti D, Vacca A, et al. Multiple myeloma exosomes establish a favourable bone marrow microenvironment with enhanced angiogenesis and immunosuppression. J Pathol. (2016) 239:162–73. doi: 10.1002/path.4712
125. Colombo M, Mirandola L, Chiriva-Internati M, Basile A, Locati M, Lesma E, et al. Cancer cells exploit notch signaling to redefine a supportive cytokine milieu. Front Immunol. (2018) 9:1823. doi: 10.3389/fimmu.2018.01823
126. De Veirman K, Wang J, Xu S, Leleu X, Himpe E, Maes K, et al. Induction of miR-146a by multiple myeloma cells in mesenchymal stromal cells stimulates their pro-tumoral activity. Cancer Lett. (2016) 377:17–24. doi: 10.1016/j.canlet.2016.04.024
127. Wang J, Hendrix A, Hernot S, Lemaire M, De Bruyne E, Van Valckenborgh E, et al. Bone marrow stromal cell-derived exosomes as communicators in drug resistance in multiple myeloma cells. Blood. (2014) 124:555–66. doi: 10.1182/blood-2014-03-562439
128. Leaf RK, Stroopinsky D, Pyzer AR, Kruisbeek AM, van Wetering S, Washington A, et al. DCOne as an allogeneic cell-based vaccine for multiple myeloma. J Immunother. (2017) 40:315–22. doi: 10.1097/CJI.0000000000000185
129. Di Noto G, Chiarini M, Paolini L, Mazzoldi EL, Giustini V, Radeghieri A, et al. Immunoglobulin free light chains and GAGs mediate multiple myeloma extracellular vesicles uptake and secondary nfkappaB nuclear translocation. Front Immunol. (2014) 5:517. doi: 10.3389/fimmu.2014.00517
130. Vulpis E, Cecere F, Molfetta R, Soriani A, Fionda C, Peruzzi G, et al. Genotoxic stress modulates the release of exosomes from multiple myeloma cells capable of activating NK cell cytokine production: Role of HSP70/TLR2/NF-kB axis. Oncoimmunology. (2017) 6:e1279372. doi: 10.1080/2162402X.2017.1279372
131. Chesi M, Matthews GM, Garbitt VM, Palmer SE, Shortt J, Lefebure M, et al. Drug response in a genetically engineered mouse model of multiple myeloma is predictive of clinical efficacy. Blood. (2012) 120:376–85. doi: 10.1182/blood-2012-02-412783
132. Martin SK, Diamond P, Gronthos S, Peet DJ, Zannettino AC. The emerging role of hypoxia, HIF-1 and HIF-2 in multiple myeloma. Leukemia. (2011) 25:1533–42. doi: 10.1038/leu.2011.122
133. Kawano Y, Kikukawa Y, Fujiwara S, Wada N, Okuno Y, Mitsuya H, et al. Hypoxia reduces CD138 expression and induces an immature and stem cell-like transcriptional program in myeloma cells. Int J Oncol. (2013) 43:1809–16. doi: 10.3892/ijo.2013.2134
134. Azab AK, Hu J, Quang P, Azab F, Pitsillides C, Awwad R, et al. Hypoxia promotes dissemination of multiple myeloma through acquisition of epithelial to mesenchymal transition-like features. Blood. (2012) 119:5782–94. doi: 10.1182/blood-2011-09-380410
135. Zahoor M, Westhrin M, Aass KR, Moen SH, Misund K, Psonka-Antonczyk KM, et al. Hypoxia promotes IL-32 expression in myeloma cells, and high expression is associated with poor survival and bone loss. Blood Adv. (2017) 1:2656–66. doi: 10.1182/bloodadvances.2017010801
136. Umezu T, Tadokoro H, Azuma K, Yoshizawa S, Ohyashiki K, Ohyashiki JH. Exosomal miR-135b shed from hypoxic multiple myeloma cells enhances angiogenesis by targeting factor-inhibiting HIF-1. Blood. (2014) 124:3748–57. doi: 10.1182/blood-2014-05-576116
137. Hu J, Van Valckenborgh E, Xu D, Menu E, De Raeve H, De Bruyne E, et al. Synergistic induction of apoptosis in multiple myeloma cells by bortezomib and hypoxia-activated prodrug TH-302, in vivo and in vitro. Mol Cancer Ther. (2013) 12:1763–73. doi: 10.1158/1535-7163.MCT-13-0123
138. Gorgun GT, Whitehill G, Anderson JL, Hideshima T, Maguire C, Laubach J, et al. Tumor-promoting immune-suppressive myeloid-derived suppressor cells in the multiple myeloma microenvironment in humans. Blood. (2013) 121:2975–87. doi: 10.1182/blood-2012-08-448548
139. Wang Z, Zhang L, Wang H, Xiong S, Li Y, Tao Q, et al. Tumor-induced CD14+HLA-DR (-/low) myeloid-derived suppressor cells correlate with tumor progression and outcome of therapy in multiple myeloma patients. Cancer Immunol Immunother. (2015) 64:389–99. doi: 10.1007/s00262-014-1646-4
140. Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, et al. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med. (2006) 203:2691–702. doi: 10.1084/jem.20061104
141. Noonan KA, Ghosh N, Rudraraju L, Bui M, Borrello I. Targeting immune suppression with PDE5 inhibition in end-stage multiple myeloma. Cancer Immunol Res. (2014) 2:725–31. doi: 10.1158/2326-6066.CIR-13-0213
142. Ray A, Tian Z, Das DS, Coffman RL, Richardson P, Chauhan D, et al. A novel TLR-9 agonist C792 inhibits plasmacytoid dendritic cell-induced myeloma cell growth and enhance cytotoxicity of bortezomib. Leukemia. (2014) 28:1716–24. doi: 10.1038/leu.2014.46
143. Ramakrishnan V, Kumar S. PI3K/AKT/mTOR pathway in multiple myeloma: from basic biology to clinical promise. Leuk Lymphoma. (2018) 59:2524–34. doi: 10.1080/10428194.2017.1421760
144. Opperman KS, Vandyke K, Clark KC, Coulter EA, Hewett DR, Mrozik KM, et al. Clodronate-liposome mediated macrophage depletion abrogates multiple myeloma tumor establishment in vivo. Neoplasia (New York NY). (2019) 21:777–87. doi: 10.1016/j.neo.2019.05.006
145. Teixidó J, Martínez-Moreno M, Díaz-Martínez M, Sevilla-Movilla S. The good and bad faces of the CXCR4 chemokine receptor. Int J Biochem Cell Biol. (2018) 95:121–31. doi: 10.1016/j.biocel.2017.12.018
146. Li X, Yao W, Yuan Y, Chen P, Li B, Li J, et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut. (2017) 66:157–67. doi: 10.1136/gutjnl-2015-310514
147. Tu MM, Abdel-Hafiz HA, Jones RT, Jean A, Hoff KJ, Duex JE, et al. Inhibition of the CCL2 receptor, CCR2, enhances tumor response to immune checkpoint therapy. Commun Biol. (2020) 3:720. doi: 10.1038/s42003-020-01441-y
148. Gutiérrez-González A, Martínez-Moreno M, Samaniego R, Arellano-Sánchez N, Salinas-Muñoz L, Relloso M, et al. Evaluation of the potential therapeutic benefits of macrophage reprogramming in multiple myeloma. Blood. (2016) 128:2241–52. doi: 10.1182/blood-2016-01-695395
149. Chen H, Li M, Sanchez E, Soof CM, Bujarski S, Ng N, et al. JAK1/2 pathway inhibition suppresses M2 polarization and overcomes resistance of myeloma to lenalidomide by reducing TRIB1, MUC1, CD44, CXCL12, and CXCR4 expression. Br J Haematol. (2020) 188:283–94. doi: 10.1111/bjh.16158
150. Vonderheide RH. CD40 agonist antibodies in cancer immunotherapy. Annu Rev Med. (2020) 71:47–58. doi: 10.1146/annurev-med-062518-045435
151. Byrne KT, Betts CB, Mick R, Sivagnanam S, Bajor DL, Laheru DA, et al. Neoadjuvant selicrelumab, an agonist CD40 antibody, induces changes in the tumor microenvironment in patients with resectable pancreatic cancer. Clin Cancer research: an Off J Am Assoc Cancer Res. (2021) 27:4574–86. doi: 10.1158/1078-0432.CCR-21-1047
152. Sun J, Chen Y, Lubben B, Adebayo O, Muz B, Azab AK. CD47-targeting antibodies as a novel therapeutic strategy in hematologic Malignancies. Leukemia Res Rep. (2021) 16:100268. doi: 10.1016/j.lrr.2021.100268
153. Tamura H, Ishibashi M, Yamashita T, Tanosaki S, Okuyama N, Kondo A, et al. Marrow stromal cells induce B7-H1 expression on myeloma cells, generating aggressive characteristics in multiple myeloma. Leukemia. (2013) 27:464–72. doi: 10.1038/leu.2012.213
154. Gorgun G, Samur MK, Cowens KB, Paula S, Bianchi G, Anderson JE, et al. Lenalidomide enhances immune checkpoint blockade-induced immune response in multiple myeloma. Clin Cancer Res. (2015) 21:4607–18. doi: 10.1158/1078-0432.CCR-15-0200
155. Ribrag V, Avigan DE, Green DJ, Wise-Draper T, Posada JG, Vij R, et al. Phase 1b trial of pembrolizumab monotherapy for relapsed/refractory multiple myeloma: KEYNOTE-013. Br J Haematol. (2019) 186:e41–e4. doi: 10.1111/bjh.15888
156. Lesokhin AM, Ansell SM, Armand P, Scott EC, Halwani A, Gutierrez M, et al. Nivolumab in patients with relapsed or refractory hematologic Malignancy: preliminary results of a phase ib study. J Clin Oncol. (2016) 34:2698–704. doi: 10.1200/JCO.2015.65.9789
157. Badros A, Hyjek E, Ma N, Lesokhin A, Dogan A, Rapoport AP, et al. Pembrolizumab, pomalidomide, and low-dose dexamethasone for relapsed/refractory multiple myeloma. Blood. (2017) 130:1189–97. doi: 10.1182/blood-2017-03-775122
158. Mateos M-V, Blacklock H, Schjesvold F, Oriol A, Simpson D, George A, et al. Pembrolizumab plus pomalidomide and dexamethasone for patients with relapsed or refractory multiple myeloma (KEYNOTE-183): a randomised, open-label, phase 3 trial. Lancet Haematol. (2019) 6:e459–e69. doi: 10.1016/S2352-3026(19)30110-3
159. Usmani SZ, Schjesvold F, Oriol A, Karlin L, Cavo M, Rifkin RM, et al. Pembrolizumab plus lenalidomide and dexamethasone for patients with treatment-naive multiple myeloma (KEYNOTE-185): a randomised, open-label, phase 3 trial. Lancet Haematol. (2019) 6:e448–e58. doi: 10.1016/S2352-3026(19)30109-7
160. Costa F, Das R, Kini Bailur J, Dhodapkar K, Dhodapkar MV. Checkpoint inhibition in myeloma: opportunities and challenges. Front Immunol. (2018) 9:2204. doi: 10.3389/fimmu.2018.02204
161. Carlsten M, Korde N, Kotecha R, Reger R, Bor S, Kazandjian D, et al. Checkpoint inhibition of KIR2D with the monoclonal antibody IPH2101 induces contraction and hyporesponsiveness of NK cells in patients with myeloma. Clin Cancer Res. (2016) 22:5211–22. doi: 10.1158/1078-0432.CCR-16-1108
162. Chan WK, Kang S, Youssef Y, Glankler EN, Barrett ER, Carter AM, et al. A CS1-NKG2D bispecific antibody collectively activates cytolytic immune cells against multiple myeloma. Cancer Immunol Res. (2018) 6:776–87. doi: 10.1158/2326-6066.CIR-17-0649
163. Yu B, Jiang T, Liu D. BCMA-targeted immunotherapy for multiple myeloma. J Hematol Oncol. (2020) 13:125. doi: 10.1186/s13045-020-00962-7
164. Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N Engl J Med. (2019) 380:1726–37. doi: 10.1056/NEJMoa1817226
165. Rodriguez-Otero P, Ailawadhi S, Arnulf B, Patel K, Cavo M, Nooka AK, et al. Ide-cel or standard regimens in relapsed and refractory multiple myeloma. N Engl J Med. (2023) 388:1002–14. doi: 10.1056/NEJMoa2213614
166. Majzner RG, Mackall CL. Clinical lessons learned from the first leg of the CAR T cell journey. Nat Med. (2019) 25:1341–55. doi: 10.1038/s41591-019-0564-6
167. Lakshman A, Kumar SK. Chimeric antigen receptor T-cells, bispecific antibodies, and antibody-drug conjugates for multiple myeloma: An update. Am J Haematol. (2022) 97:99–118. doi: 10.1002/ajh.26379
168. Moreau P, Garfall AL, van de Donk N, Nahi H, San-Miguel JF, Oriol A, et al. Teclistamab in relapsed or refractory multiple myeloma. N Engl J Med. (2022) 387:495–505. doi: 10.1056/NEJMoa2203478
170. Lesokhin AM, Tomasson MH, Arnulf B, Bahlis NJ, Miles Prince H, Niesvizky R, et al. Elranatamab in relapsed or refractory multiple myeloma: phase 2 Magnet isMM-3 trial results. Nat Med. (2023) 29(9):2259–67. doi: 10.1038/s41591-023-02528-9
171. Dhillon S. Elranatamab: first approval. Drugs. (2023) 83(17):1621–7. doi: 10.1007/s40265-023-01954-w
172. Xia J, Li Z, Xu K. Immunotherapies targeting GPRC5D in relapsed or refractory multiple myeloma: latest updates from 2022 ASH Annual Meeting. J Hematol Oncol. (2023) 16:60. doi: 10.1186/s13045-023-01461-1
173. Zhao J, Ren Q, Liu X, Guo X, Song Y. Bispecific antibodies targeting BCMA, GPRC5D, and FcRH5 for multiple m yeloma therapy: latest updates from ASCO 2023 Annual Meeting. J Hematol Oncol. (2023) 16(1):92. doi: 10.1186/s13045-023-01489-3
174. Shrivastava T, Van Rhee F, Al Hadidi S. Targeting B cell maturation antigen in patients with multiple myeloma: current perspectives. OncoTargets Ther. (2023) 16:441–64. doi: 10.2147/OTT.S370880
175. Kazandjian D, Kowalski A, Landgren O. T cell redirecting bispecific antibodies for multiple myeloma: emergin g therapeutic strategies in a changing treatment landscape. Leuk Lymphoma. (2022) 63(13):3032–43. doi: 10.1080/10428194.2022.2113532
176. Moreaux J, Hose D, Kassambara A, Reme T, Moine P, Requirand G, et al. Osteoclast-gene expression profiling reveals osteoclast-derived CCR2 chemokines promoting myeloma cell migration. Blood. (2011) 117:1280–90. doi: 10.1182/blood-2010-04-279760
177. Delgado-Calle J, Bellido T, Roodman GD. Role of osteocytes in multiple myeloma bone disease. Curr Opin Support Palliat Care. (2014) 8:407–13. doi: 10.1097/SPC.0000000000000090
178. Lai FP, Cole-Sinclair M, Cheng WJ, Quinn JM, Gillespie MT, Sentry JW, et al. Myeloma cells can directly contribute to the pool of RANKL in bone bypassing the classic stromal and osteoblast pathway of osteoclast stimulation. Br J Haematol. (2004) 126:192–201. doi: 10.1111/j.1365-2141.2004.05018.x
179. Abe M, Hiura K, Wilde J, Shioyasono A, Moriyama K, Hashimoto T, et al. Osteoclasts enhance myeloma cell growth and survival via cell-cell contact: a vicious cycle between bone destruction and myeloma expansion. Blood. (2004) 104:2484–91. doi: 10.1182/blood-2003-11-3839
180. Raje N, Terpos E, Willenbacher W, Shimizu K, García-Sanz R, Durie B, et al. Denosumab versus zoledronic acid in bone disease treatment of newly diagnosed multiple myeloma: an international, double-blind, double-dummy, randomised, controlled, phase 3 study. Lancet Oncol. (2018) 19:370–81. doi: 10.1016/S1470-2045(18)30072-X
181. Fulciniti M, Tassone P, Hideshima T, Vallet S, Nanjappa P, Ettenberg SA, et al. Anti-DKK1 mAb (BHQ880) as a potential therapeutic agent for multiple myeloma. Blood. (2009) 114:371–9. doi: 10.1182/blood-2008-11-191577
182. Iyer SP, Beck JT, Stewart AK, Shah J, Kelly KR, Isaacs R, et al. A Phase IB multicentre dose-determination study of BHQ880 in combination with anti-myeloma therapy and zoledronic acid in patients with relapsed or refractory multiple myeloma and prior skeletal-related events. Br J Haematol. (2014) 167:366–75. doi: 10.1111/bjh.13056
183. Azab AK, Runnels JM, Pitsillides C, Moreau AS, Azab F, Leleu X, et al. CXCR4 inhibitor AMD3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy. Blood. (2009) 113:4341–51. doi: 10.1182/blood-2008-10-186668
184. Ramonell KM, Zhang W, Hadley A, Chen CW, Fay KT, Lyons JD, et al. CXCR4 blockade decreases CD4+ T cell exhaustion and improves survival in a murine model of polymicrobial sepsis. PloS One. (2017) 12:e0188882. doi: 10.1371/journal.pone.0188882
185. Han AR, Lee JY, Kim HJ, Min WS, Park G, Kim SH. A CXCR4 antagonist leads to tumor suppression by activation of immune cells in a leukemia-induced microenvironment. Oncol Rep. (2015) 34:2880–8. doi: 10.3892/or.2015.4297
186. Santagata S, Napolitano M, D'Alterio C, Desicato S, Maro SD, Marinelli L, et al. Targeting CXCR4 reverts the suppressive activity of T-regulatory cells in renal cancer. Oncotarget. (2017) 8:77110–20. doi: 10.18632/oncotarget.v8i44
187. Chen Y, Ramjiawan RR, Reiberger T, Ng MR, Hato T, Huang Y, et al. CXCR4 inhibition in tumor microenvironment facilitates anti-programmed death receptor-1 immunotherapy in sorafenib-treated hepatocellular carcinoma in mice. Hepatology. (2015) 61:1591–602. doi: 10.1002/hep.27665
188. Ghobrial IM, Liu CJ, Redd RA, Perez RP, Baz R, Zavidij O, et al. A phase ib/II trial of the first-in-class anti-CXCR4 antibody ulocuplumab in combination with lenalidomide or bortezomib plus dexamethasone in relapsed multiple myeloma. Clin Cancer Res. (2020) 26:344–53. doi: 10.1158/1078-0432.CCR-19-0647
189. Koldehoff M, Beelen DW, Elmaagacli AH. Small-molecule inhibition of proteasome and silencing by vascular endothelial cell growth factor-specific siRNA induce additive antitumor activity in multiple myeloma. J Leukoc Biol. (2008) 84:561–76. doi: 10.1189/jlb.0907632
190. Markovina S, Callander NS, O'Connor SL, Xu G, Shi Y, Leith CP, et al. Bone marrow stromal cells from multiple myeloma patients uniquely induce bortezomib resistant NF-kappaB activity in myeloma cells. Mol Cancer. (2010) 9:176. doi: 10.1186/1476-4598-9-176
191. Prince HM, Honemann D, Spencer A, Rizzieri DA, Stadtmauer EA, Roberts AW, et al. Vascular endothelial growth factor inhibition is not an effective therapeutic strategy for relapsed or refractory multiple myeloma: a phase 2 study of pazopanib (GW786034). Blood. (2009) 113:4819–20. doi: 10.1182/blood-2009-02-207209
192. Somlo G, Lashkari A, Bellamy W, Zimmerman TM, Tuscano JM, O'Donnell MR, et al. Phase II randomized trial of bevacizumab versus bevacizumab and thalidomide for relapsed/refractory multiple myeloma: a California Cancer Consortium trial. Br J Haematol. (2011) 154:533–5. doi: 10.1111/j.1365-2141.2011.08623.x
193. White D, Kassim A, Bhaskar B, Yi J, Wamstad K, Paton VE. Results from AMBER, a randomized phase 2 study of bevacizumab and bortezomib versus bortezomib in relapsed or refractory multiple myeloma. Cancer. (2013) 119:339–47. doi: 10.1002/cncr.27745
194. Podar K, Zimmerhackl A, Fulciniti M, Tonon G, Hainz U, Tai YT, et al. The selective adhesion molecule inhibitor Natalizumab decreases multiple myeloma cell growth in the bone marrow microenvironment: therapeutic implications. Br J Haematol. (2011) 155:438–48. doi: 10.1111/j.1365-2141.2011.08864.x
195. Dabbah M, Jarchowsky-Dolberg O, Attar-Schneider O, Tartakover Matalon S, Pasmanik-Chor M, Drucker L, et al. Multiple myeloma BM-MSCs increase the tumorigenicity of MM cells via transfer of VLA4-enriched microvesicles. Carcinogenesis. (2020) 41:100–10. doi: 10.1093/carcin/bgz169
196. Hu J, Handisides DR, Van Valckenborgh E, De Raeve H, Menu E, Vande Broek I, et al. Targeting the multiple myeloma hypoxic niche with TH-302, a hypoxia-activated prodrug. Blood. (2010) 116:1524–7. doi: 10.1182/blood-2010-02-269126
Keywords: bone marrow microenvironment, cellular compartments, noncellular compartments, multiple myeloma, treatment
Citation: Lu K, Wang W, Liu Y, Xie C, Liu J and Xing L (2024) Advancements in microenvironment-based therapies: transforming the landscape of multiple myeloma treatment. Front. Oncol. 14:1413494. doi: 10.3389/fonc.2024.1413494
Received: 07 April 2024; Accepted: 20 June 2024;
Published: 17 July 2024.
Edited by:
Mario I. Vega, University of California, Los Angeles, United StatesReviewed by:
Caterina Riillo, Magna Græcia University of Catanzaro, ItalyCopyright © 2024 Lu, Wang, Liu, Xie, Liu and Xing. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lijie Xing, eGlhb3BpYW80MjNAMTI2LmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.